00:02:52.640
Hi everyone, welcome back after lunch! I hope you enjoyed your meal. Right now, we are going to start discussing our topic today: high-speed parsing of massive XML data in Ruby. I am Tetsuya Hirota from Japan.
00:03:24.560
Today, I'll tell you more about high-speed processing of XML in Ruby. This presentation is an updated version of one I previously delivered at Ruby World Conference 2023 in Japan. First, I was requested to convert data in the BioProject XML file into JSON using Ruby.
00:04:10.760
While I was provided with a sample call to do this, the process took me more than four times longer than the sample code suggested. Therefore, I created a method similar to the Python `iterparse()`, which parsed each top-level element individually. This method, combined with Ruby on Ractor, improved performance to be about 30% faster than the original Python sample.
00:05:06.600
Today's content will include an introduction, a brief self-introduction, an overview of my company, and an explanation of my project. I will cover what I attempted to achieve and how I created a mechanism similar to Python's `iterparse()` using Ruby.
00:05:36.560
My name is Tetsuya Hirota, and I am the CTO at my company. I'm based in Japan, where I spend a lot of time developing applications for bioinformatics. I have been dedicated to our core biotic database development for quite some time.
00:06:10.760
In my work, I also teach a programming course focusing on lifelong learning related to bioinformatics. I offer valuable insights into life sciences, including data visualization to help understand what types of operations are demanded by the life science community.
00:07:02.640
Today, my focus will be on ongoing development processes for bioinformatics databases. We face challenges related to high-speed parsing of massive XML files.
00:07:25.200
The BioProject is a public database that encompasses various bioscience projects. This includes information related to genomes and RNA sequences registered in public databases, along with associated metadata such as papers, experimental conditions, and samples.
00:07:38.000
My project involves curating a metacollection about sequence analysis with robust metadata. The format of this project is primarily simple XML files that exceeded 2 gigabytes in size, containing over 700,000 records. The database is continuously growing.
00:08:13.800
Researchers often need to perform complex queries on this data. The current limitations mean heavy memory use, consuming around 70 gigabytes when parsing the files.
00:08:49.720
Initially, I attempted to parse the BioProject XML using standard methods, which resulted in non-responsive outputs that took over 35 minutes and consumed excessive memory.
00:09:05.640
This process is slow because it necessitates loading all objects into memory, and each request searches through the entire dataset. My goal has been to find a more efficient method.
00:09:49.720
I explored Python sample codes, which process data in about 8 minutes through incremental loading of individual elements.
00:10:10.240
The elements in our database are structured hierarchically with each project element averaging between 100 kilobytes to a few megabytes in size. The XML parser traditionally extracts all elements into memory, creating significant overhead.
00:11:04.360
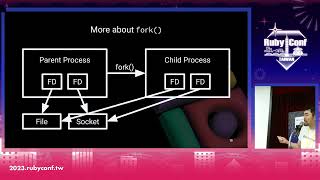
To improve this process, I began leveraging string operations to separate data intelligently and utilize memory more efficiently. Initially, I focused on Ruby's existing capabilities but ran into performance issues, illustrating the importance of implementing solutions that avoid blocking processes.
00:12:01.720
I decided to split these tasks into more manageable parts. In the first part, I extracted the primary XML data, followed by the parsing of secondary elements, ensuring that the output was streamlined.
00:12:45.920
For outputting JSON, I gathered each project element and processed it before writing it to the resulting file.
00:13:08.680
This modular approach allows for much quicker results, reducing processing time significantly.
00:13:43.280
Utilizing Ruby on Ractor has enabled concurrent processing, which I found beneficial in optimizing the performance compared to Ruby's conventional parsing methods.
00:14:41.560
In the end, I was able to achieve a processing time of about five minutes and thirty-four seconds, which was a significant improvement from the initial methods I tried.
00:15:29.560
Moreover, during these tests, I utilized a five CPU setup with 8 gigabytes of RAM, showcasing how resource allocation impacts performance.
00:16:09.440
In conclusion, my approach to bioinformatics database processing in Ruby can be optimized greatly. It is essential that we take advantage of Ruby's unique features to sidestep potential bottlenecks.
00:17:38.840
I appreciate everyone's attention and I’m ready for any questions you might have.
00:31:10.720
Thank you for your interest and participation today! If you have any further queries about the technology or my methodologies, I welcome you to ask.